Research
I am a computational molecular and integrative biologist and mathematical scientist. Currently, my research is in three areas:
- Massively parallel characterization of CRISPR specificity.
- State of the art indel-correcting DNA barcodes.
- Bat transcriptomics, phylogenetic history, and positive selection.
CRISPR Specificity: CHAMP
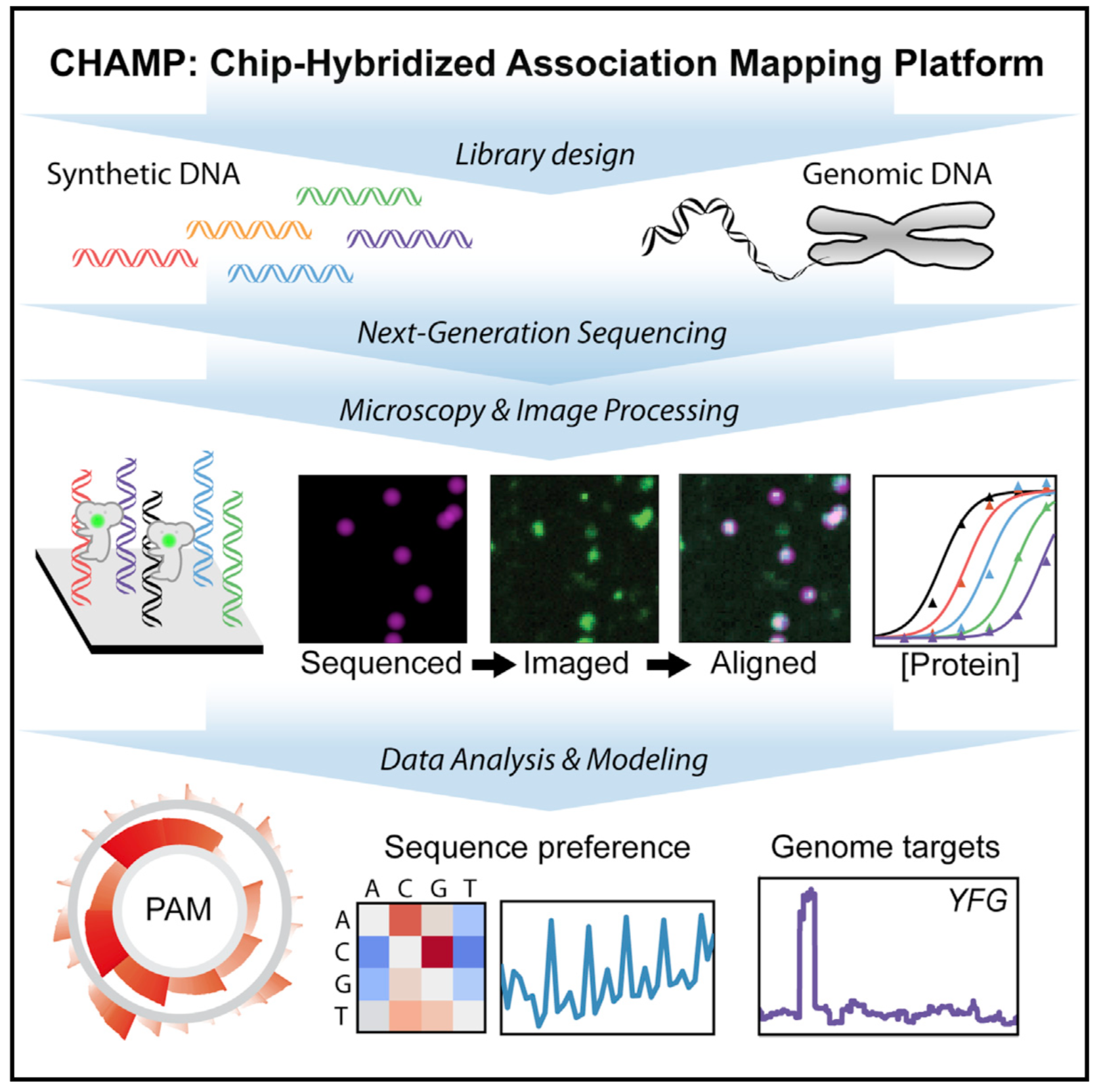
The past few years have seen a revolution in gene-editing technology thanks to the discovery and development of the CRISPR family of proteins for gene-editing. But wild type CRISPR proteins are known to occasionally cut DNA in unintended locaitons, which could potentially cause new diseases in patients. Before CRISPR can gain wide use in human medicine, we need better understanding and control of its DNA targeting to ensure it does exactly and only what we want it to.
I worked with a team from the Finkelstein lab to develop the Chip-Hybridized Association Mapping Platform (CHAMP), a massively parallel platform for characterizing protein-DNA binding interactions. It works by recovering Illumina sequencing chips after sequencing, performing further biochemical assays on the sequenced chip, and observing reactions with a total internal reflection fluorescence (TIRF) microscope. I used Fourier methods, linear least squares, machine learning, and non-linear optimization to turn large-scale raw data into per-sequence binding specificity information.
We used this platform to interrogate the target specificity of the most common CRISPR system in nature, the CRISPR Cascade complex. I visualized the binding specificity of several classes of sequences and developed mathematical and statistical models to characterize the Cascade complex specificity profile. We fully characterized an extended 6 bp protospacer adjacent motif (PAM) and showed that while the canonical 3 bp are the most important, positions 4, 5, and 6 can have a significant effect on intermediate PAMs. We also found a novel periodic reduction of specificity every 3 + 6n bases in the target sequence. We were then able to use our method to look at the binding specificity for a CRISPR protein across an entire human exome for the first time. This technique can be used in a clinical setting to verify in vitro the safety of a proposed gene-therapy treatment on a per-patient basis.
State of the Art Barcoding: FREE Barcodes
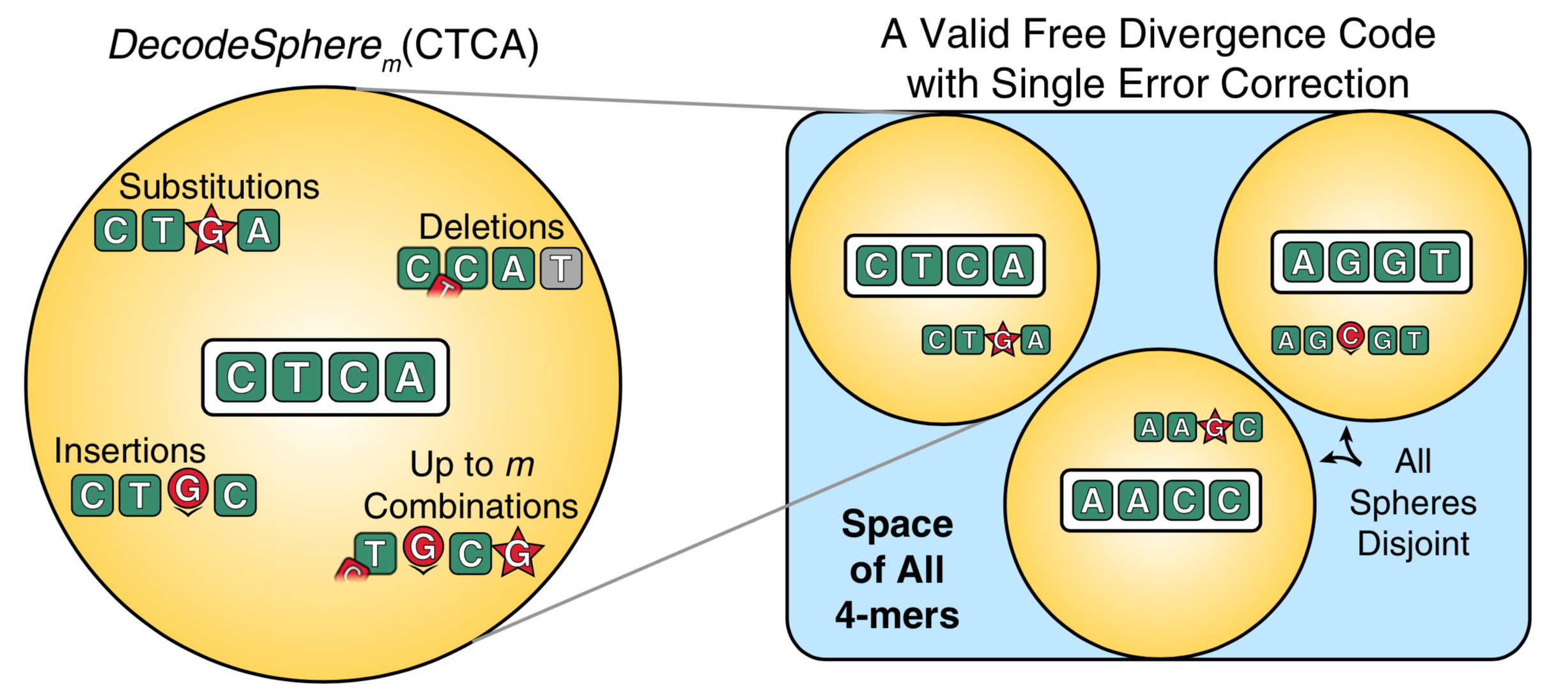
DNA barcodes, short DNA sequences used to label individual biomolecules in pooled populations, are ubiquitous throughout modern biology. This simple technology has enabled the development of exciting new assays such as single-cell RNA sequencing and massively parallel drug screening. However, DNA barcodes are subject to standard DNA errors: substitutions, insertions, and deletions. Previous methods have used Hamming or Levenshtein distance to try to correct some of these errors, but neither of these metrics correctly accounts for insertions and deletions in DNA barcodes due to uncertainty in the location of the right end of the barcode.
I developed and experimentally validated Filled/truncated Right End Edit (FREE) barcodes to correctly account for the kinds of errors observed in real DNA barcodes. This required the development of new high-dimensional algorithms, finite field linear algebra, dynamic programming, and the proof of various properties of FREE divergence space. Because FREE barcodes correctly account for the kinds of errors seen in DNA barcodes, they are able to correct errors with higher accuracy than previous methods, using fewer bp for the same number of barcodes. I generated a single-error correcting barcode library with >106 unique barcodes. And due to their construction, FREE barcodes can be easily concatenated. This allows for combinatorially large barcode libraries and/or arbitrarily low error rates with different strategies. I demonstrated these possibilities with an example library with >1015 barcodes and an example library with error rates of <10-19.
Bat Phylogenetics and Positive Selection
The order Chiroptera, the order of bats, is one of the most interesting and diverse orders of mammals. Not only are they interesting due to their ability to fly and echolocate, but they are of interest to human health because they are the known or suspected reservoirs of several deadly human viruses, including SARS, Nipah, Hendra, and Ebola. Improving our understanding of bat cellular pathways can give us insight into how these viruses function in their natural hosts, leading the way for more effectively combatting these diseases.
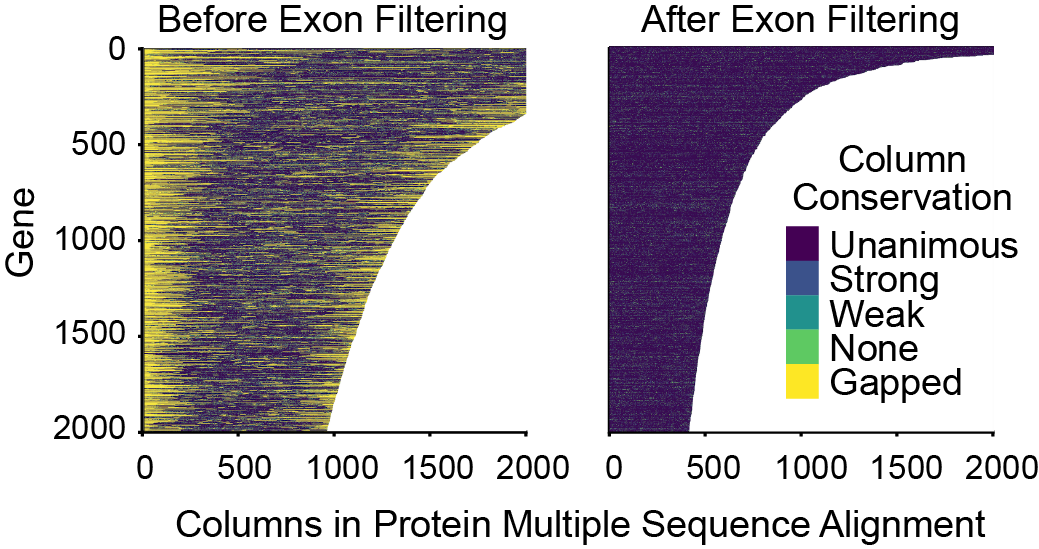
I performed an meta-analysis of publically available DNA sequencing data and novel data we sequenced ourselves to perform order-wide phylogenetic and positive selection analyses. To improve results of these analyses based on data from heterogenous public data sources, I developed a new statistical method called MIXR, Mismatching Isoform eXon Remover, for detecting and removing non-random alignment artifacts. Positive selection, indicated by a dN/dS statistic value >1, is a signature of genes which have a history of viral antagonism. We identified 181 such genes, which will be of interest for further investigation by the virology community. My collaborators have, in fact, already used the sequences of one gene family, NPC1, to identify one of the key amino acids in the bat NPC1 protein which acts as a barrier to cell entry and infection from the Ebola virus.